U.S. Food and Drug Administration (FDA)

The U.S. Food and Drug Administration (FDA) is responsible for ensuring the safety, effectiveness, and quality of pharmaceuticals, biologicals, and medical devices intended for human use as well as the safety of food, cosmetics, and radiation-emitting products. In addition, this agency is responsible for the safety, efficacy, and quality of pharmaceuticals and feed intended for animal use. As of 2009, this agency is also responsible for regulating the sale of tobacco products.
Food
The FDA’s Center for Food Safety and Applied Nutrition (CFSAN) regulates food and food additives sold in the United States. CFSAN’s Redbook 2000 is a manual of test methods that the agency expects will be performed for novel food additives or ingredients. These tests include the use of many animals and multiple species; some typically involve dogs (or puppies) and, in some cases, last one year.
CFSAN also requires the monitoring of shellfish for algal toxins that accumulate in their flesh and can cause health problems in humans. For decades, this monitoring meant injecting liquefied shellfish into the abdominal cavities of mice—an excruciating process that causes convulsions, severe pain, paralysis, and, ultimately, death when a shellfish is contaminated.
PETA regulatory scientists have worked closely with both the European Commission and the FDA to replace this test for all types of shellfish hazards.
Drugs
The marketing of drugs and other pharmaceutical products in the U.S. is controlled by the Federal Food, Drug, and Cosmetic Act (FFDCA), which empowers the FDA’s Center for Drug Evaluation and Research to require extensive toxicity testing on animals before a new drug is deemed “safe” for marketing. In some instances, the FDA calls for additional testing on animals after products have been approved. For example, despite decades of data supporting the safety of sunscreen products, the agency demands that sunscreen manufacturers test the active ingredients in their products on animals.
In order to satisfy FDA data requirements, thousands of rats, mice, rabbits, dogs, and primates are killed in preclinical laboratory toxicity tests to assess the safety of new drugs (including all ingredients and even minor differences in formulation).
Commonly required animal tests include the following:
- Acute (short-term) toxicity: 7 to 20 rats + dogs or primates
- Subchronic (14 to 180 days) toxicity: rats + dogs or primates
- Chronic (lifetime) toxicity: 120 rats + 32 dogs or primates
- Cancer-causing effects: 400 rats + 400 mice
- Toxicity to reproductive systems
- Segment I (reproductive toxicity in 2 generations): 2,500 rats
- Segment II (birth defects): 900 rabbits + 1,300 rats
- Segment III (peri- and postnatal effects): rats
- Absorption, distribution, metabolism, excretion, and pharmacological interactions of active ingredients
- Specialty studies
- Genetic toxicity: 80 hamsters/mice x 2 to 5 separate studies
- Immune system toxicity: 32 rats
- Skin/eye/mucosal irritation: 3 rabbits per test
Following this extensive battery of animal testing, drugs generally undergo three phases of clinical trials before they are considered for widespread human use. The fact that months or years of human studies are required over and above the standard battery of animal tests suggests that health authorities do not trust the results of animal experiments—and for good reason. A significant number of drugs are rejected during human clinical trials because they are found to cause toxic and other adverse health effects “not predicted” in preclinical animal experiments.
In fact, the National Institutes of Health reports that 95 out of every 100 drugs that successfully pass animal trials and go into human clinical testing fail during the human clinical trial phase.
The problem is that species differences are so vast that animal results are, at best, a very poor approximation of what will happen in humans or, at worst, dangerously misleading. The alternative is to advance science to the point where preclinical tests are based on human biology, which will better predict what will happen to real human volunteers or patients in the clinical trials.
Today, cutting-edge technology can allow us to better predict complex human reactions.
Tobacco
The marketing of tobacco products in the U.S. is controlled by the FFDCA as amended by the Family Smoking Prevention and Tobacco Control Act (FSPTCA), which is administered by FDA’s Center for Tobacco Products (CTP). In contrast to other products that the FDA regulates, tobacco products are inherently unsafe. Nevertheless, the FSPTCA makes the FDA responsible for ensuring that any new tobacco product entering the market is “appropriate for the protection of the public health.” This is possible if a new product reduces risk for tobacco users without increasing risk for non-users (for example, by encouraging non-users to initiate tobacco use). While the FSPTCA names only cigarettes, smokeless tobacco, and roll-your-own tobacco, it also gives the FDA authority to deem other tobacco products subject to the Act. As of August 8, 2016, any product meeting the statutory definition of a tobacco product is subject to the Act, including hookah, e-cigarettes, dissolvables, cigars, and pipe tobacco, as well as future tobacco products.
In its guidance to industry on submitting marketing applications for new tobacco products, FDA includes in vivo (animal) tests among the types of tests that might be used when comparing risk between products. As a result, PETA is concerned that animal testing of tobacco products will increase. Because e-cigarettes and other electronic nicotine delivery systems (ENDS) are new products that may present a reduced health risk to users compared to conventional cigarettes, PETA is especially concerned that animal tests will be conducted in support of marketing applications for these products.
PETA has submitted public comments on the FDA’s regulation of tobacco products, including its deeming rule and guidance to industry on both modified risk tobacco product applications and premarket applications for ENDS. PETA has also met directly with CTP to urge the agency to accept only data from nonanimal tests. In its final deeming rule, the FDA addressed each of PETA’s recommendations, such as setting product standards to reduce the need for new testing. In its recent draft guidance on premarket applications for ENDS products, the FDA now encourages applicants to meet with CTP early in the development process to discuss the suitability of nonanimal tests and provides clear guidance on literature reviews, computational modeling and certain in vitro tests. These important first steps will reduce the number of animals used to test tobacco products. PETA will continue to participate in the regulatory process until no animals are used.
PETA regulatory scientists also work with industry and academia to promote the development and acceptance of nonanimal methods. For example, human lung tissue, either donated or reconstructed from cultured cells, can be exposed to cigarette smoke or e-cigarette vapor directly. Such methods are more relevant to how people actually use tobacco products than forcing rats or dogs to inhale these substances before killing and dissecting them. In 2015, a PETA researcher coauthored a review of recent publications describing the use of nonanimal methods to study tobacco products, and PETA has also participated in workshops organized by the Institute for In Vitro Sciences to promote these methods.
The FSPTCA does not specifically require animal tests, so it is within the FDA’s authority to require that only nonanimal methods be used to support new products. Internationally, Belgium, Estonia, Germany, Slovakia, and the United Kingdom have all banned testing tobacco products using animals demonstrating that this is an achievable goal.
Biologicals
“Biologicals” are medicinal products, such as vaccines, hormones, antibodies, and blood products, that are derived from living organisms. In the U.S., biologicals are regulated by the FDA’s Center for Biologics Evaluation and Research. Because of their origin, biologicals must undergo extensive quality control during production.
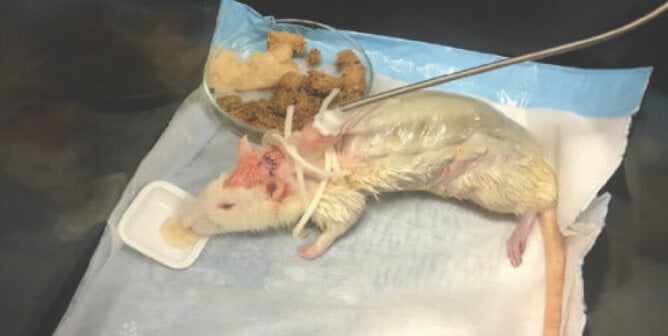
Vaccine testing in particular consumes an estimated 2.5 million animals every year because vaccines are often produced by weakening, inactivating, or detoxifying a virulent microorganism or toxin. Each batch of the finished product is then tested on animals, causing them pain, suffering, and death.
Safety testing is carried out to try to make sure that a safe immune response is observed and that people who are inoculated with the vaccine are not infected by the pathogen. A common study is the “abnormal toxicity test,” in which guinea pigs and mice are injected with a biological product and observed for one week. The test may be repeated multiple times for the same product until all of the following criteria are met:
- All the animals survive the observation period.
- None of the animals show any weight loss at the end of the observation period.
- None of the animals show toxic signs.
Numerous other disease-specific animal safety tests have also been developed, such as the “mouse weight-gain test,” which is used for the whole-cell pertussis vaccine. In this test, mice injected with the vaccine are observed for weight gain and to see whether they are alive or dead after 72 hours and again after one week.
Another test, for the oral polio vaccine, called the “neurovirulence test,” is also devastating to animals. In this test, rhesus or cynomolgus monkeys receive an injection of the vaccine in their spines, are observed for up to three weeks for signs of paralysis, and are then killed and examined.
“Potency testing” is carried out to determine the effectiveness of inactivated (nonliving) vaccines in protecting the recipient against bacterial or viral infections. These studies use “challenge” tests, in which large numbers of animals—usually mice, rats, guinea pigs, rabbits, and/or chickens—are inoculated with a vaccine and then “challenged” through purposeful infection with the disease that the vaccine is designed to protect against.
To test the potency of a single batch of rabies vaccine, for example, live rabies virus is injected through the skulls and directly into the brains of 160 mice. Some of these mice are given the protective vaccine first, but some are not. These cranial injections are extremely painful and completely irrelevant to the normal route of infection. Approximately half of the animals develop and/or die of rabies, a painful neurological disease involving tremors, loss of control over one’s body, the inability to swallow, and severe weight loss. Analytical methods have been developed by rabies vaccine manufacturers, but they are not yet validated or accepted for regulatory use.
Medical Devices
The FDA’s Center for Devices and Radiological Health is responsible for the licensing of medical devices, which include an extremely broad range of products, such as cardiac pacemakers, stents, insulin pumps, and surgical devices.
A great deal of animal testing for these products relates to the safety of the materials in the medical devices: the plastic polymers, the metals, and the ceramics. These materials are implanted into an animal’s tissues, or chemicals leached from these products are injected into their tissues to observe the toxicity to animals. Researchers are generally looking for toxicity to cells and allergic potential, as well as applying the acute and chronic toxicity tests described above (in the “Drugs” section). These “biocompatibility tests” (click here for more information) are often conducted on mice, rats, and rabbits. Similar to what is done with biologics, each batch of a medical device is also tested for quality control using animals.
Testing the effectiveness of medical devices is often difficult because these devices are specifically designed for human anatomy and physiology. Pigs, sheep, and dogs are the most commonly used species for efficacy testing of these devices, even though the structure of the animals’ bodies is often too dissimilar to be of use.
Physicians and medical device sales specialists also use animals (primarily dogs and pigs) during training exercises in which the animals are often implanted with the device and later killed.
Non-animal methods include human cadavers, bench-top simulators of various human organs, cell-based tests, and computer programs that can simulate, for example, the effect of a particular heart rhythm as encoded into a pacemaker.

Please send a short, polite comment to the TFDA thanking them for proposing the new draft regulations and urging them to finalize them so no more animals die for blood sugar or blood lipid tests.

Sign the petition urging the FDA to immediately update its requirements to allow manufacturers to readily use an animal-free test method.

The comment period is now closed. Thank you to all who took action! We’ll keep you updated. In the meantime, please urge companies in Taiwan to stop experimenting on and killing animals for marketing food and beverage products. Original post: After ongoing talks with PETA and other animal protection groups, the Taiwan government has proposed … Read more »
Urge Secretary of War Pete Hegseth to cut waste, fraud, and abuse by banning the Pentagon from funding foreign experiments on animals.

The bill will finally stop the intense suffering monkeys have endured for generations at the hands of U.S. experimenters and importers. Take action to help!

The parent company of one of Hong Kong’s most popular online shopping platforms is severing animals’ heads and limbs in dozens of deadly experiments.


